Summary
Is amyloid-β harmful to the brain? The literature is replete with evidence that individuals who harbour amyloid-β show a number of neurological effects that are similar to those seen in Alzheimer's disease. While studies are sometimes contradictory, there is substantial evidence that amyloid-β is associated with cross-sectional and progressive brain atrophy, cross-sectional network dysfunction and longitudinal cognitive decline. A closer examination of the data indicates that simply classifying individuals based on the presence or absence of amyloid-β does not give the full picture. Studies that have categorized normal older people according to preclinical stages of Alzheimer's disease strongly indicate that those with evidence of both amyloid-β and biomarkers suggesting neurodegeneration show the greatest evidence of decline. Thus examining individuals simply in terms of the presence or absence of amyloid-β is not as informative as defining where they stand along a putative pathway of preclinical Alzheimer's disease progression.
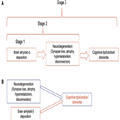
Figure 1.
Two different conceptualizations of how amyloid-β affects neurological outcomes. A summarizes a preclinical staging scheme for Alzheimer's disease in which amyloid-β deposition is the initiating event. In this scheme, brain amyloid-β deposition alone constitutes Stage 1 of preclinical Alzheimer's disease, followed by Stage 2 in which amyloid-β leads to neurodegeneration. In Stage 3, subtle cognitive dysfunction insufficient to establish dementia or mild cognitive impairment, occurs. This scheme posits neurodegeneration as the invariable mediator between amyloid-β and cognition. B suggests an alternative view in which neurodegeneration and amyloid-β are independent processes. Neurodegeneration without amyloid-β has been referred to as suspected non-Alzheimer pathology (SNAP). In this scheme, either neurodegeneration or amyloid-β alone may lead to cognitive dysfunction, although the two together may produce synergistic harmful effects.
Many of these studies, especially those that are cross-sectional, demonstrate associations but do not prove causality. Scientific inquiry, especially in human research, benefits from studies of association especially when results converge across different methods and laboratories. However, traditional views of the scientific method hold that disproving a hypothesis is more valuable than accumulating positive evidence. In this regard, the only true human experiments manipulating amyloid-β levels—clinical trials of amyloid-lowering therapies—refute the importance of amyloid-β in Alzheimer's disease. This has not, however, disproven the amyloid hypothesis primarily because it is not a true hypothesis but rather a complex model of disease causation. Over time, this model has been refined and generated numerous hypotheses, one of which is that Alzheimer's disease has a prolonged incubation period during which amyloid-β promotes neurological damage. Failure of clinical trials in late stage disease is fully compatible with this model and begs for studies to be done in early and presymptomatic individuals. The limitations of cross-sectional association studies are in part ameliorated through longitudinal studies that permit observation of the evolution of disease from asymptomatic stages. While incomplete, the studies reviewed here show that amyloid-β deposition is a very early event that appears to play a harmful role in brain ageing especially when it is associated with neurodegeneration.
In this setting, the appropriate question, therefore, may not be whether amyloid-β is harmful to the brain, but how amyloid-β is harmful to the brain. The mechanisms that link amyloid-β to neurodegeneration are poorly understood and perplexing. For example, in Alzheimer dementias with focal features such as posterior cortical atrophy or progressive aphasia, the pattern of amyloid-β deposition does not reflect the pattern of brain degeneration (Rabinovici et al., 2008; Rosenbloom et al., 2011). Individuals may respond differently to amyloid-β deposition based on the ability to compensate (Elman et al., 2014b) or the presence of genetic factors that influence immunity and inflammation (Tanzi, 2015). Multiple biomarkers must be measured to characterize neurodegeneration, which could be important for 'staging' individuals by defining how progressed they are—an important factor for selection of individuals in clinical trials. The addition of tau imaging offers the potential of more specific biomarkers for degenerative brain processes (Villemagne et al., 2015) that may shed light on these issues.
Cross-sectional association studies also have the disadvantage that they reflect brain biomarkers of structure and function that do not necessarily reflect neurodegeneration but could indicate a variety of non-progressive processes. The time between the appearance of brain amyloid-β and the development of dementia is potentially decades (Villemagne et al., 2013) during which a multitude of pathological mechanisms may be operating. We need longitudinal studies to define relationships between amyloid-β, neurodegeneration, and cognition, and to better infer causation from temporal associations. How often is neurodegeneration preceded by amyloid-β, and how often might it arise independently and interact with amyloid-β to result in decline? Is Fig. 1A or Fig. 1B closer to the evolution of Alzheimer's disease? In Fig. 1A, dementia occurs via amyloid-β-induced neurodegeneration, while in Fig. 1B dementia could evolve in relation to either amyloid-β or neurodegeneration independently. This is one of the key controversies in Alzheimer's disease research today, but it is not simply an academic exercise. Our ability to target the right interventions to the correct processes at the appropriate time point depends on a more precise understanding of the complex chain of events that occurs over many years to produce dementia.
No comments:
Post a Comment